近日,中南大学生命科学学院医学遗传学研究中心邬玲仟、李卓课题组在《CellDeath &Disease》(细胞死亡与疾病)发表题为“PIGK defects induce apoptosis in Purkinje cells and acceleration of neuroectodermal differentiation(PIGK功能缺陷导致浦肯野细胞凋亡和神经外胚层分化加速)”的研究论文,揭示了PIGK功能丧失导致磷脂酰肌醇生物合成缺陷22型(GlycosylphosphatidylinositolBiosynthesisDefect 22,GPIBD22)的致病机制。博士研究生陈思仪为论文第一作者,邬玲仟、李卓和梁德生教授为共同通讯作者。该研究受国家重点研发计划项目、国家自然科学基金等项目支持。

磷脂酰肌醇生物合成缺陷症(GPIBD)是一类严重的致愚、致死性遗传病,患者主要表现为全面发育迟缓、智力障碍、癫痫发作、肌张力低下、进行性脑萎缩等神经发育障碍症状,部分患者还存在特殊面容、骨骼肢体发育异常。PIGK双等位基因突变可导致GPIBD22型,但目前PIGK功能缺陷导致GPIBD22的病理和分子致病机制尚不清楚。为此,邬玲仟、李卓团队在前期发表了首个中国人群GPIBD22家系遗传病因研究的基础上,进一步通过构建首个条件性Pigk敲除小鼠模型以及PIGK功能缺陷iPSC模型,阐明了细胞凋亡增加、神经祖细胞分化异常和浦肯野细胞进行性丢失是GPIBD22患者神经发育异常的细胞病理基础;同时验证了未折叠蛋白反应(UPR)过度激活是GPIBD22主要分子致病机制之一,并通过添加UPR通路抑制剂Kira6可缓解病变发展。该研究为GPIBD这类严重遗传病的预防和诊治提供了新见解。
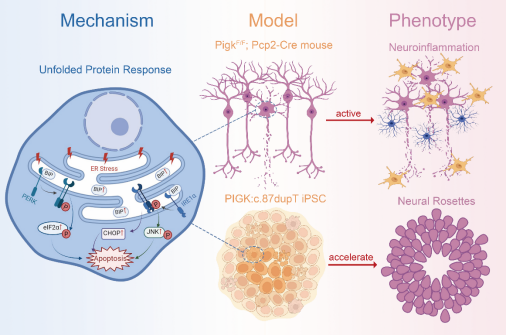
图1.PIGK 功能缺陷导致GPIBD22的致病机制示意图
邬玲仟、梁德生、李卓教授带领的临床遗传学团队,长期专注于遗传病的临床诊疗、致病机制研究,以及防控技术的开发与转化应用,相关工作获得了多项国家重点研发计划和国家自然科学基金等项目资助。