
Title: Haplotype-resolved long-read sequencing reveals parent-of-origin effects of tandem-repeat variation in autism spectrum disorder
Jinchen Li , Tengfei Luo , Xue Ren , Songyu Yang , Hui Quan , Yan Zhu , Senwei Tan , Xiangbin Jia , Pei Yu , Yuanfeng Huang , Zhaowei Jiang , Xudong Xiang , Hailiang Guo , Tengfei Zhu , Zhiqing Hu , Miaojin Zhou , Qian Zhang , Yanchen Li , Hanwen Gong , Ruiting Liu , Meining He , Xun Zhou , Dianang Han , Zhikun Wang , Ting Bai , Xingxing Jian , Guihu Zhao , Bin Li , Yidong Shen , Jianjun Ou , Qian Pan , Tianyun Wang , Hui Guo 5, Zhengmao Hu , Beisha Tang , Lu Xia , Kun Xia
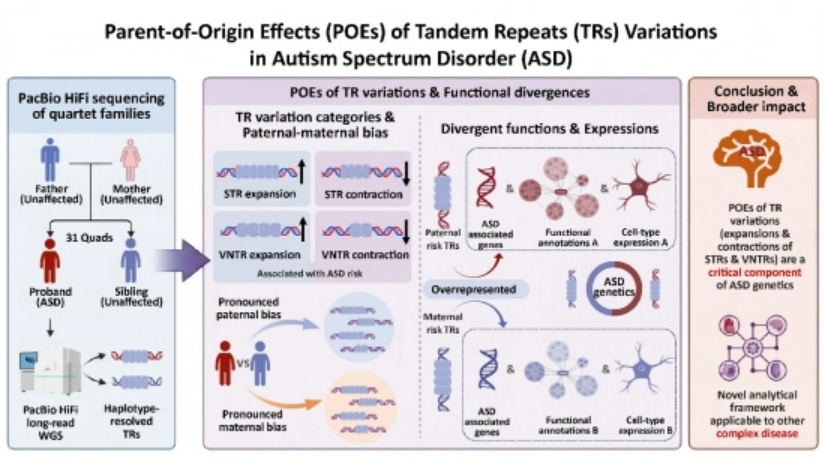
Autism Spectrum Disorder (ASD) is a complex neurodevelopmental disorder, whose core clinical manifestations include social communication deficits, restricted interests, and repetitive stereotyped behaviors. ASD has a heritability as high as 80%, indicating that genomic variations serve as a key etiological factor of ASD. Studies have demonstrated that approximately 10–20% of ASD cases can be explained by copy number variations and de novo mutations, while a considerable proportion of the genetic risk remains unelucidated. Tandem Repeats (TRs) are widely distributed in the human genome and represent one of the major sources of genomic variation. Based on the length of repeat units, TRs are classified into Short Tandem Repeats (STRs) with repeat units of 2–6 bp, and Variable Number Tandem Repeats (VNTRs) with repeat units of 7–100 bp. However, constrained by the read length limitations of first- and second-generation sequencing technologies, previous studies have only focused on the correlation between STR expansion and ASD. The associations of STR contraction, VNTR expansion and contraction, as well as their parental origin effects with ASD remain largely unclear. Based on the Autism Clinical and Genetic Resources in China (ACGC) cohort, this study enrolled 31 four-person pedigrees (including ASD-diagnosed probands, unaffected siblings, fathers and mothers), covering a total of 124 samples. Whole-genome PacBio HiFi long-read sequencing was performed, and the repeat counts of over one million STRs and VNTRs across the whole genome of each sample were quantified at the haplotype level. The results revealed that ASD probands exhibited significantly more abnormal contraction and expansion events of STRs and VNTRs compared with their unaffected siblings. Moreover, aberrant variation patterns of different types of TRs showed a prominent paternal or maternal parental preference. Further analyses indicated that although paternally and maternally derived risk TRs were both enriched in known ASD-related genes, they displayed distinct enrichment signatures in genomic regulation, functional pathways, and cell-type-specific expression patterns. This study suggests that abnormal TRs of different parental origins may affect genes with diverse functions through distinct regulatory mechanisms, thereby increasing the risk of ASD. It provides a novel perspective for understanding the vital role of TR variations in complex diseases.
Link:https://doi.org/10.1016/j.scib.2026.03.058





 No.172, Tongzipo Road,Changsha City,Hunan
No.172, Tongzipo Road,Changsha City,Hunan  0731-89665629
0731-89665629  life_csu@163.com
life_csu@163.com 